



 Rna Folding: predicting secondary structure
Rna Folding: predicting secondary structure

Life depends for many essential processes on RNA (RiboNucleic Acid). Roughly speaking, each RNA molecule is a unique sequence of many nucleotides based on 4 different nucleotides. This is called, the primary RNA structure. This sequence determines how the RNA functions, but not exclusively so. It was discovered that RNAs are not loosely floating strings in the cell, but that in each RNA various parts glue together, forming a uniquely structured bun; see picture at the right. Such a bun consists of structures known as hairpins, internal loops, bulges, multiple loops. This so-called secondary structure (and the tertiary structure based on it) highly influences the functioning of the RNA.
The most frequently used approach to determine the secondary structure of RNA are minimal energy algorithms (see for example MFOLD by Zuker. Such programs require a lot of computer power (speed as well as memory), and predict roughly 3/4 of the secondary structure correctly.
In a joint project with the Institute of Molecular Biology we programmed 3 completely
different algorithms:
1. greedy algorithm, 2. stochastic algorithm and 3. genetic algorithm.
We used the program language APLbecause this is a powerful language which
enables us to develop and maintain a complex program in reasonable short time.
Being an interpreter, the resulting code may not be as fast as code from a compiled language.
Nevertheless we could predict the structure on small PC's in a very reasonable time
(in the eighties it took a few hours on a standard computer of that time to predict the structure for a RNA of 300 nucleotides).
For shorthand we called this program STAR (STructure Analysis of Rna).
Doing a simulation will sometimes (although not always) yield a better prediction than minimal energy predictions. Moreover it gives information about the folding process, rather than exclusively a prediction of "the" final folding. Doing such a simulation allows us to study the RNA dynamics. For example we can investigate the folding pathway of the RNA molecule during its synthesis, thereby predicting intermediate foldings and in some cases functionally important metastable states. And it may show some alternative foldings that are maybe only slightly less probable and could emerge when circumstances are a bit different.
Most programs use the minimal-energy-algorithm to predict RNA structure. Unfortunately, such an algorithm can not predict pseudoknots. However, our three algorithms can predict pseudoknots.
What is a pseudoknot?
A pseudoknot is a very special way of folding.
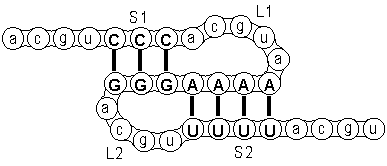 First, the RNA string folds back upon itself, thereby forming a so-called hairpin.
First, the RNA string folds back upon itself, thereby forming a so-called hairpin.
The figure on the right shows this first hairpin-structure where three nucleotides CCC
pair with GGG forming a loop L1 and a stem S1.
Next, some of the nucleotides in the remaining sequence form a second coupling with nucleotids in the loop.
You can see this in the figure where nucleotides AAAA in the first loop pair with UUUU,
thereby forming a second loop L2 and a second stem S2.
This is the most basic type of pseudoknot.
A simple but lucid way to present such a pseudoknot is by "bracket-view":
acguCCCacguaAAAAGGGacguUUUUacgu
::::(((:::::[[[[)))::::]]]]::::
Here pairing is shown by associated open- and close- brackets of similar type.
Pairs of stem S1 are shown by ((())) and pairs of stem S2 by
[[[[]]]].
More complex pseudoknots require more than 2 different types of brackets.
See for example the following pseudoknot:

The secondary and tertiary structure of RNA is extremely important. Knowing this helps in understanding the function of certain RNA's and also the non-function of related (mutated) sequences. It can explain phylogenetic similarities and dissimilarities; it can explain why some parts are easily mutated and others are not; and so on.
So the two main advantages of our program STAR are that 1) it simulates the folding pathway and 2) it can predict pseudoknots.
 History of STAR
History of STAR
The main "engines" of STAR were created by three people: Jan Pieter Abrahams, Sacha Gultyaev and me, in close coöperation with Cees Pley (from the Leiden institute of Chemistry).
Jan Pieter Abrahams ignited the first creative spark. In 1985 he worked as a student for me when he developed the Greedy Algorithm. This program could predict the secondary structure of sequences of 100 to 200 nucleotides in one or two hours on an Atari personal computer, contrary to minimum energy algorithms that needed much more time on more powerful computers at that time. Furthermore this greedy algorithm could predict pseudoknots, a feat that minimal energy algorithms could not do.
Sacha Gultyaev developed the Stochastic Algorithm first in 1990 when he was working at the Institute of Influenza in St.Petersburg, Russia. Coming to Theoretical Biology at Leiden University in 1993, he perfected this program and reprogrammed it in APL for inclusion in program STAR.
In 1994 I developed the first version of the Genetic Algorithm. This worked in a rather simple way and Sacha Gultyaev refined it to the sophisticated program that it is nowadays.
Several students contributed to STAR when working on projects for me.
Mirjam v.d. Berg ported STAR to Atari ST and JeanJack Riethoven to PC-DOS.
Victor Bos wrote most of the reference manual and extended the menu-options.
Monique Weijnen and John Voskuilen worked on the user interface.
Finally, several bug-removals, speed improvement and polishing of
the user interface were done by Viktor Hopmans and Jacky Ng.
They also finished and tested the port of STAR from Macintosh to Windows.
Although I take full responsibility for the final program it is obvious that I have to thank many others for their contributions.
 STAR references
STAR references
Our work on STAR is published in various articles. If you are interested we can recommend the following publications:
- The first article about STAR and presenting the greedy algorithm was:

Abrahams J.P., van den Berg M., van Batenburg E. & Pleij C.W.A. (1990):
Prediction of RNA secondary structure, including pseudoknotting, by computer simulation. Nucl. Acids Res. 18, 3035-3044. - The stochastic algorithm is presented in:
Gultyaev A.P. (1991):
The computer simulation of RNA folding involving pseudoknot formation. Nucl. Acids Res. 19, 2489-2494. - Articles which explain the genetic algorithm are:
van Batenburg F.H.D., Gultyaev A.P. and Pleij C.W.A. (1995):
An APL-programmed Genetic Algorithm for the Prediction of RNA Secondary Structure. J. theor. Biol. 174, 269-280.
Gultyaev A.P., van Batenburg F.H.D. and Pleij C.W.A. (1995):
The Computer Simulation of RNA Folding Pathways Using a Genetic Algorithm. J. Mol. Biol. 250, 37-51. - A few examples of scientific work where STAR was used:
- Bringloe,D.H., Gultyaev,A.P., Pelpel,M., Pleij,C.W.A. and Coutts,R.H.A. (1998):
- The nucleotide sequence of satellite tobacco necrosis virus strain C and helper-assisted replication of wild-type and mutant clones of the virus. J.of General Virology 79,1539-1546.
- Gultyaev,A.P., Batenburg,F.H.D.van and Pleij,C.W.A. (1999):
- An approximation of loop free energy values of RNA H-pseudoknots. RNA 5 609-617.
- Gultyaev,A.P., Batenburg,F.H.D.van and Pleij,C.W.A. (1998):
- Dynamic competition between alternative structures in viroid RNAs simulated by an RNA folding algorithm. J.Mol.Biol. 276 43-55.
- Gultyaev,A.P., Franch,T. and Gerdes,K. (1997):
- Programmed cell death by hok/sok of plasmid R1: coupled nucleotide covariations reveal a phylogenetically conserved folding pathway in the hok family of mRNAs. J.Mol.Biol. 273, 26-37.
- Gultyaev,A.P., Batenburg,F.H.D.van and Pleij,C.W.A. (1995):
- The influence of a metastable structure in plasmid primer RNA on antisense RNA binding kinetics. Nucleic Acids Res. Vol.23, No 18, 3718-3725.
- Gultyaev,A.P., Batenburg,F.H.D.van and Pleij,C.W.A. (1994):
- Similarities between the secondary structure of satellite tobacco mosaic virus and tobamovirus RNAs. J.of General Virology 75, 28511-2856.
- Hellendoorn,K., Mat,A.W., Gultyaev,A.P. and Pleij,C.W.A. (1996):
- Secondary structure model of the coat protein gene of Turnip Yellow Mosaic Virus RNA: lon, C-rich, single stranded regions. Virology 224, 43-54.
- Jeeninga,R.E., Huthoff,H.T., Gultyaev,A.P. and Berkhout,B. (1998):
- The mechanism of actinomycin D-mediated inhibition of HIV-1 reverse transcription. Nucleic Acids Res. Vol.26,no.23, 5472-5479.
- Leone,G., Schijndel,H.van, Gemen,B.van, Kramer,F.R. and Schoen,C.D. (1998):
- Molecular beacon probes combined with amplification by NASBA enable homogeneous, real time detection of RNA. Nucleic Acids Res. Vol.26,no.9, 2150-2155.
- Proutski,V., Gaunt,M.W., Gould,E.A. and Holmes,E.C. (1997):
- Secondary structure of the 3'-untranslated region of yellow fever virus: implications for virulence, attenuation and vaccine development. J.of General Virology 78, 1543-1549.
- Proutski V., Gould E.A. and Holmes E.C. (1997a):
- Secondary structure of the 3' untranslated region of flaviviruses: similarities and differences. Nucleic Acids Res. Vol. 25, No. 6, 1194-1202.
- Rodriguez-Alvarado G. and Roossinck M.J.(1997b):
- Structural Analysis of a Necrogenic Strain of Cucumber Mosaic Cucumovirus Satellite RNA in Planta. Virology 236, 155-166.
 STAR algorithms
STAR algorithms
STAR supports 3 algorithms to predict the secondary structure of an RNA sequence.
All algorithms can predict H-pseudoknots.
Furthermore, all three algorithms compute the folding pathway,
rather than the final structure prediction only.
This pathway is a very rough one in the Greedy algorithm,
the Genetic algorithm produces the most subtle and realistic pathway; the pathway
computed by the Stochastic algorithm is somewhat in between.
The Greedy Algorithm
The Greedy Algorithm simulates the formation of a RNA structure by adding one stem at a time to the growing structure. Each step, the stem that adds most to the stability of the structure of that point is incorporated.
A true simulation should take both the rate of formation and the rate of destruction of stems into account. For the sake of simplicity the Greedy Algorithm presumes that each intermediate structure is well defined and more or less stable.
In this way you can describe the addition of stems to the intermediate structure as a series of linked equilibria. As soon as a local equilibrium is reached, STAR seeks the next most favorable one.
- References:
- Abrahams J.P., van den Berg M., van Batenburg E. & Pleij C.W.A. (1990): Prediction of RNA secondary structure, including pseudoknotting, by computer simulation. Nucl. Acids Res. 18, 3035-3044.
The Stochastic Algorithm
The Stochastic Algorithm folds RNA through a procedure of stepwise improving local equilibrium structures.
Similarly to the Greedy Algorithm, the Stochastic
Algorithm does not (explicitly) take disruption of stems into account.
The essential difference is in the way to add stems to the structure,
that is, to determine the stem most likely to form at any subsequent folding step.
In the Greedy Algorithm each step adds the stem with the greatest energy contribution.
This ignores the possibility that a particular ensemble of stems could
add more to the stability of structure than the stem with the best energy gain.
The Stochastic Algorithm tests if some stem combinations are better than the (energetically) best stem. Thus this algorithm applies a Monte Carlo procedure. At every step, the addition of stems to the final structure starts with the generation of a "population" of randomized structures. This population uses the most stable stems that are compatible with the previously formed (intermediate) structure.
Next stems are added with probabilities depending on their energy values. The probability to add particular stems is proportional to kinetic parameter K: energy gain divided by destabilizing energy of loop.
After the random structure generation, the best structures are taken. Those stems that are common in all are added to the final structure. The addition uses a priority of descending order for kinetic parameter K.
In the next steps this whole procedure is repeated. Newly formed random structures add new stems that are compatible with the previously formed (intermediate) structure.
Furthermore, the algorithm can simulate RNA synthesis during folding. This is done by starting the folding in a small initial part at the 5´ end of the RNA chain and "grow" by increasing this string with small increments. "Growth" increments the chain at the end of iterations when the number of stems is compatible with those previously included to structure is small.
- References:
- Gultyaev A.P. (1991). The computer simulation of RNA folding involving pseudoknot formation. Nucl. Acids Res. 19, 2489-2494.
The Genetic Algorithm
"Genetic Algorithm" is the name for a special type of searching algorithm. The name derives from the fact that the procedure simulates the fitness-determined competition between individuals of a population, similarly to the process of natural evolution.
In our application, the individuals of the population are the folded structures that evolve during the simulation. The main advantage of this procedure is that it considers the competition of structures rather than that of possible stems (like in the Greedy Algorithm and the Stochastic Algorithm). This allows disruption of some stems in intermediate steps. Thus the algorithm simulates RNA folding pathway in some "energy-landscape" which overcomes energy barriers by stem disruption and descends into energy minima by stem formation. Generally, this seems to be the best way to simulate the RNA folding process.
At any point in the simulation, the Genetic Algorithm deals with a "population" of structures, which are produced by a randomized procedure, somewhat similar to the Stochastic Algorithm. Initially the population contains only unfolded states. Every Genetic Algorithm iteration (the unit of the main program cycle) produces some new structures by changing previous ones and selecting the fittest solutions, thus yielding a new population with the same number of structures.
The selection procedure usually improves the fitness (free energy) of structures in the population, finally producing relatively stable structures.
The changes in structures are "Genetic Algorithm mutations" (disruption as well as formation of some stems) and "Genetic Algorithm crossovers" (the randomized generation of a new structure by combining stems from several parental solutions into compatible combination).
The probabilities of stem addition and disruption depend on energy contributions of stems: the more stable the stem, the greater the probability to include it to the structure and the less the probability to remove it.
Every structure in the population is mutated once and the crossover includes all stems of all current structures. So a population of N structures produces a temporary population of 2N+1 possible structures according to the fitness (free energy).
This completes one Genetic Algorithm iteration. This procedure is repeated again until no improvement is found.
The Genetic Algorithm also performs "growth" in a manner which is similar to that option in the Stochastic Algorithm. The folding of the uncompleted RNA chain is simulated by starting with a small part at the 5´ end of the chain (about 10 nucleotides). The chain length increases with increments of which the size depends on the free energy of structures in current population.
- References:
- van Batenburg F.H.D., Gultyaev A.P. and Pleij C.W.A. (1995): An APL-programmed Genetic Algorithm for the Prediction of RNA Secondary Structure. J. theor. Biol. 174, 269-280.
- Gultyaev A.P., van Batenburg F.H.D. and Pleij C.W.A. (1995): The Computer Simulation of RNA Folding Pathways Using a Genetic Algorithm. J. Mol. Biol. 250, 37-51.
Output Views
STAR supports several ways of viewing the calculated secondary structure.
STAR does not provide a graphical view to see your structure.
For this we included "ConnecT View" which can be used in other programs
(such as RNAdraw and LoopDLoop) for a graphical display and
Bracket View can be used by PseudoViewer
for graphical display.
Those 7 views are...
Table View

The first four columns specify the location of the stem:
column 1 - the base number of most 5´ nucleotide of the stem,
column 2 - base number of the 3´ end of the 5´ stem-half,
column 3 - the base number of the 5´ end of the 3´ stem-half,
column 4 -the base number of the most 3´ nucleotide of the
stem.
The next 3 columns specify the energy content of the stem:
column 5 - Gibbs Free Energy of the base pairs in kcal/mol,
column 6 - Gibbs Free Energy of the single stranded stretch of RNA
located directly between the corresponding bases of the second and third column in kcal/mol,
column 7 - Gibbs Free Energy that the stem contributes to the structure of previous stems;
each value is the sum of column 5 and 6 unless there is a loop outside
the stem; the sum of column 7 yields the total energy of the whole
structure.
Span View
Span view output of the previous table looks like:
You see at the right a stem with a big bulge UAAUUUUCAGCACCAGGG. At the left you see a pseudoknot with stem GCC-GGU (5-7;15-17) and stem GCCUU-AGGGU (10-14;21-25).
Above the sequence you see the stems drawn.
Span view draws a stem by connecting two single stranded stemhalfs.
Like this:

Lego View
Lego view output looks like:

Lego View is more compact than the Mountain View and displays the same information.
This view was a modification that I developed from the Mountain view.
We annotated the figure to show you what a loop, a stem and a bulge looks like in lego view.
N.B. This view displays pseudoknots incorrectly!
Mountain View
Mountain view output looks like:
Pairing nucleotides are connected by horizontal lines; the center is indicated by a ´|´. So slopes are the stems and loops are the plateaus. We annotated the figure to show you what a loop, a stem and a bulge looks like.
This display was first created by Hogeweg & Hesper.
- References:
- P.Hogeweg and Hesper,B. (1984). Energy directed folding of RNA sequences. Nucleic Acids Research Vol.12, 67-74.
N.B. This view displays pseudoknots incorrectly!
Bracket View
Bracket view output looks like:
The first row, starting with the $-sign, shows the nucleotides.
The second row, following the %-sign, shows the pairing: opening parenthesis that match the
next-nearest closing parenthesis shows a stem.
We annotated the figure to show you how a loop, a stemhalf and a pseudoknot looks.
Pseudoknots are very obvious by the use of brackets instead of parentheses.
Bracketview output can be used to visualise the structure with PseudoViewer. You [Copy] bracketview from STAR and [Paste] it in the structure-window of PseudoViewer but you should remove the number and the "$" and the "%" in front of each line.
ConnecT View

STAR only provides character-type of output rather then graphical output. For this reason ConnecT View is implemented to migrate the predicted structure to drawing programs such as RNAdraw and LoopDLoop.
N.B. The majority of drawing programs (including RNAdraw and LoopDLoop) CANNOT handle pseudoknots and will give unpredictable results (or rather "the predictable" result of a crash).
Currently the best program to visualise the RNA structure, including pseudoknots, is PseudoViewer. This program uses (a modified version of) bracket-view for input.
Path View
Path view output looks like:
Path View shows the steps with the added and removed stems. You see the result from calculating the secondary structure with the Genetic Algorithm.
This pathview shows the dynamics of the folding. It enables you to analyse the folding process rather than the final result. This pathway sometimes shows some frequently recurring structures. In our research such structures have occasionally proved to be metastable structures that were already observed in experiments.
 Download
Download
After my retirement I stopped development and support of STAR, but you can download STAR on an as-is base for free:
- windows version 5 (STARWin.zip)
- mac version 5 (STAR5Mac.zip)
- mac version 6-beta (STAR6Mac.zip) (this version can draw a graphic, but this is not fully tested).
- Greedy algorithm: Abrahams J.P., van den Berg M., van Batenburg E. & Pleij C.W.A. (1990): Prediction of RNA secondary structure, including pseudoknotting, by computer simulation. Nucl.Acids Res.18, 3035-3044.
- Stochastic algorithm: Gultyaev A.P. (1991): The computer simulation of RNA folding involving pseudoknot formation. Nucl.Acids Res.19, 2489-2494.
- Genetic algorithm: van Batenburg F.H.D., Gultyaev A.P. & Pleij C.W.A. (1995): An APL-programmed Genetic Algorithm for the Prediction of RNA Secondary Structure. J.theor.Biol.174, 269-280 (download).